
提起瑞士,你会不假思索地想到钟表。品质上乘、可与时间分秒博弈的钟表,必定要有精湛的制造工艺作为依托,除此之外,还要经得起岁月洪波的洗礼,有如中流砥柱,历久弥坚。就在这个国土面积仅有四万多平方公里的“钟表之国”,一度崛起许多久负盛名的钟表品牌:百达翡丽(Patek Philippe)、爱彼(Audemars Piguet)、江诗丹顿(Vacheron Constantin)……
▲图片来源:Pixabay
除了瑞士钟表,还有一种器械可与这个国度紧密关联——瑞士军刀。这是一种集多种实用功能于一身的折叠刀具,通常包含主刀、小刀、牙签、剪刀、改锥、开罐器、螺丝刀、镊子等十几种基本工具,使用时只需将特定的工具从刀柄中牵拉出来,便可满足现实生活中的许多需求,因而又叫作万用刀。加之外型小巧,携带方便,成为不少旅行者外出的必备品。

▲图片来源:Pixabay
“一刀多用”的思维同样启发了科学研究,化学家也想寻找一把“万用刀”,借助一种反应体系合成多类不同结构的产物。实现这一过程的基本思路是首先将底物转化为关键的活性中间体,再引入其他官能化试剂与其特定的反应位点作用。此前我们介绍过德国马克思·普朗克煤炭研究所(Max-Planck-Institut für Kohlenforschung)Tobias Ritter教授团队发展的高区域选择性构建芳基C-N键的工作(见文末推荐阅读)。他们发现,使用四氟噻蒽对芳香烃进行C-H键官能化具有十分优异的对位选择性,反应得到的芳基噻蒽鎓盐可在常规过渡金属催化剂或光氧化还原催化剂存在的情况下参与一系列的C-C键偶联反应,还可实现卤化、磺酰化、磷酸酯化、胺化等多类C-X键的构建。
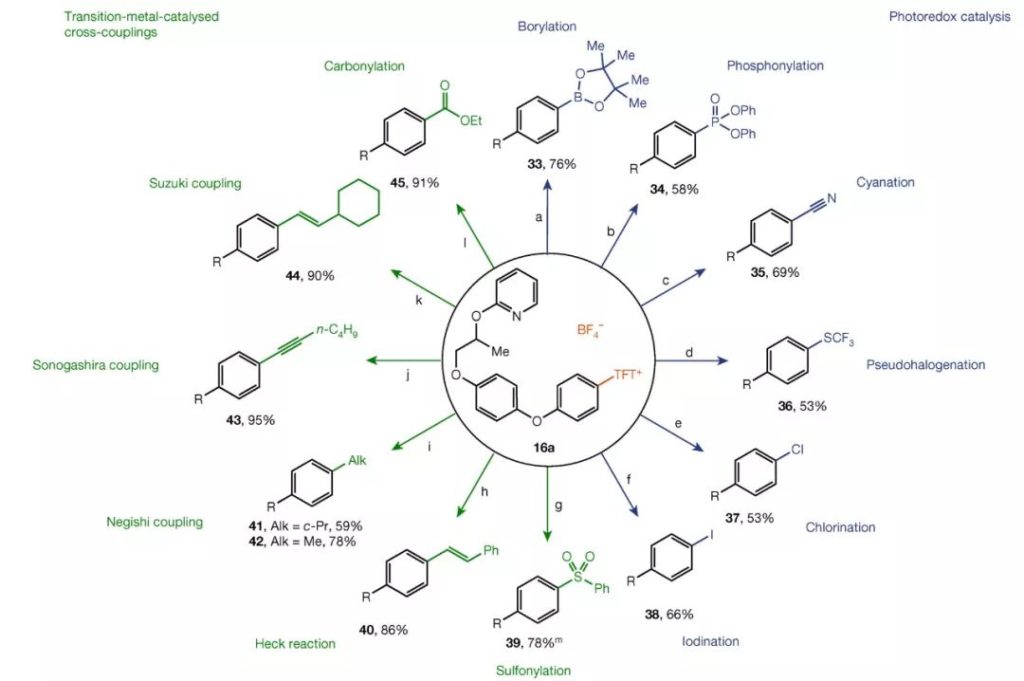
▲从芳基噻蒽鎓盐出发实现多种芳基C-C键及C-X键的构建(图片来源:参考资料[1])
除此之外,天然产物全合成研究中的汇聚式合成(convergent synthesis,也称集群式合成)也借鉴了这一思路。研究者首先从特定的原料出发构建核心分子骨架,再经由这一结构、通过不同的反应路线发散地合成一系列天然产物分子。例如,2017年,四川大学的秦勇教授团队便借助光引发的自由基串联途径快速构建了柯楠因型(Corynanthe-type)的单萜并吲哚生物碱结构,再通过后续转化完成了多达33种具有重要生理活性的天然产物分子的汇聚式合成。
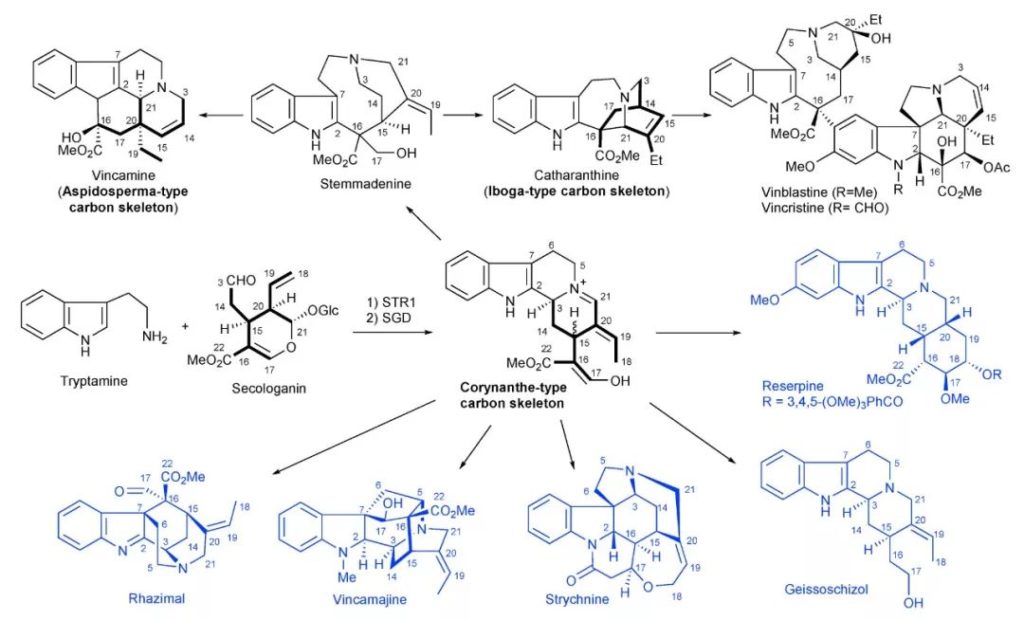 ▲从柯楠因型单萜并吲哚生物碱结构出发实现多种天然产物分子的汇聚式合成(图片来源:参考资料[2])
▲从柯楠因型单萜并吲哚生物碱结构出发实现多种天然产物分子的汇聚式合成(图片来源:参考资料[2])
最近,美国斯克里普斯研究所(The Scripps Research Institute,TSRI)的余金权教授团队同样发展了一种通用策略,只要将烷基羧酸的β-C-H键活化,转化为相应的β-丙内酯,后续便可实现其与一系列亲核试剂的亲核取代开环,得到多种不同β-C-H键官能化的产物。相关工作以同行评议(peer-reviewed)文章的形式发表在顶级学术期刊Nature上。

▲图片来源:参考资料[3]
C-H键活化反应从上世纪60年代发展至今已取得了显著的突破。反应的底物从最初修饰不可消除的强配位导向基团(DG)至可消除的弱配位DG,再到瞬态DG(transient directing group),最终发展成为无DG参与的反应。C-H键活化的位点从近程发展至远程活化,又从化学、区域选择性的探索升级为具有立体选择性的不对称C-H键活化。C-H键官能化的种类也从C-C键偶联拓展为C-O、C-N键等其他C-X键的形成过程。脂肪族羧酸在自然界中广泛存在,并且种类丰富,因而可作为廉价易得的合成砌块用于构建多种复杂的结构。不少烷基羧酸包含β-C-H键,设计C-H键官能化反应时,羧基可用作导向基团与过渡金属催化剂配位,β-C-H键与过渡金属中心作用形成金属杂五元环中间体,由此实现C-H键活化,随后进一步与其他官能化试剂反应。不过,每一类C-H键官能化反应均有其特定的底物适用范围。
以C-C键偶联为例,C-H键烷基化过程仅可使用一级烷基碘化物或烷基硼酸参与反应,二级或三级烷基化试剂的反应效果较差;C-H键烯基化仅适用于贫电子的烯烃;对于芳基化过程,活性较高的碘代芳香烃可有效发生反应,而反应活性较低的溴代与氯代芳香烃不适用于C-H键官能化。此外,如何在羧基未经保护的情况下,通过烷基羧酸β-C-H键的活化实现氟化、羟基化、胺化等形成C-X键的过程目前也尚无有效的解决方案。如此看来,烷基羧酸β-C-H键的官能化反应还有很大的提升与拓展空间。
余金权教授课题组设想出这样一种解决方案,烷基羧酸在Pd催化剂的作用下发生β-C-H键活化,形成Pd杂五元环中间体,此时体系中不加入任何官能化试剂,通过特定的驱动条件促使其直接发生还原消除,形成β-丙内酯活性中间体。这类结构的环张力较大,具有很高的反应活性,因而即便是亲核性较弱的亲核试剂,也有望与之反应得到烷基羧酸β-C-H键官能化的产物。
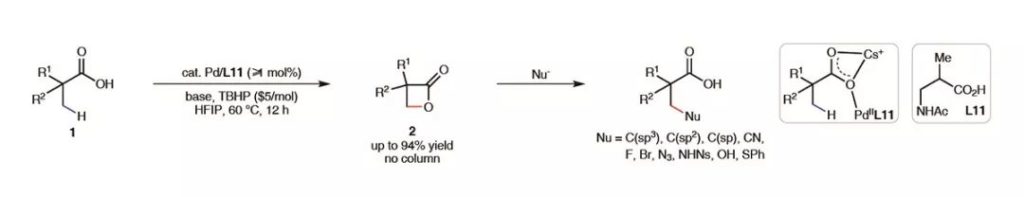
▲β-丙内酯作为关键活性中间体实现烷基羧酸β-C-H键的多种官能化(图片来源:参考资料[3])
然而,实现以上过程并不容易,难点体现在以下几个方面:(1)形成β-丙内酯活性中间体需经历四元环状过渡态,相比于γ-C-H键活化并进一步形成γ-丁内酯,前者在能量上十分不利。因而反应倾向于得到γ-丁内酯产物,以往的一些研究已充分体现了这一点;(2)相当一部分基团的亲核性比羧基强,假使分子中特定位点存在这类基团(如含氮基团),其会与过渡金属中心优先配位,最终还原消除形成更稳定的C-N键偶联产物,因此设计底物分子时还需注意在某些位点合理规避修饰相应的基团;(3)即便形成了Pd杂五元环中间体,由于β-丙内酯结构环张力大,较不稳定,该中间体很容易以开环途径发生还原消除,形成如下图所示的非环状C-O键偶联产物,常见的竞争副反应途径包括乙酰氧基化、烷氧基化等。
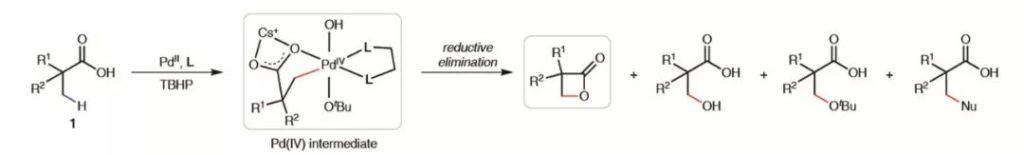 ▲Pd杂五元环中间体还原消除可能形成的副产物(图片来源:参考资料[3])
▲Pd杂五元环中间体还原消除可能形成的副产物(图片来源:参考资料[3])
作者以2,2-二甲基丁酸作为模板底物,考察不同Pd(II)预催化剂及氧化剂参与反应的情况。在大多数情况下,反应主要得到非环状的C-O键偶联产物。随后他们尝试了以空间位阻较大的叔丁基过氧化氢(TBHP)作为氧化剂,Pd(CH3CN)2Cl2作为Pd(II)预催化剂,体系中有少量β-丙内酯产物形成,并且没有观察到γ-丁内酯及其他开环副产物。TBHP将Pd(II)盐氧化为Pd(IV)盐后,形成的–OtBu、OH–对Pd(IV)中心具有很强的配位能力,很难发生还原消除,可最大程度抑制以上副反应过程。另外,–OtBu空间位阻较大,可加速其他配体还原消除,因而还能促进目标产物的形成。
他们进一步考察了不同配体对反应效果的影响。合适的配体可能会加速β-丙内酯产物的形成,双齿螯合的配体与过渡金属中心形成六元环比形成五元环的咬合角更大,更有利于其他配体的还原消除。作者最终发现,α位甲基取代的N-乙酰基-β-丙氨酸(L11)作为配体参与反应时可以得到最佳的结果。
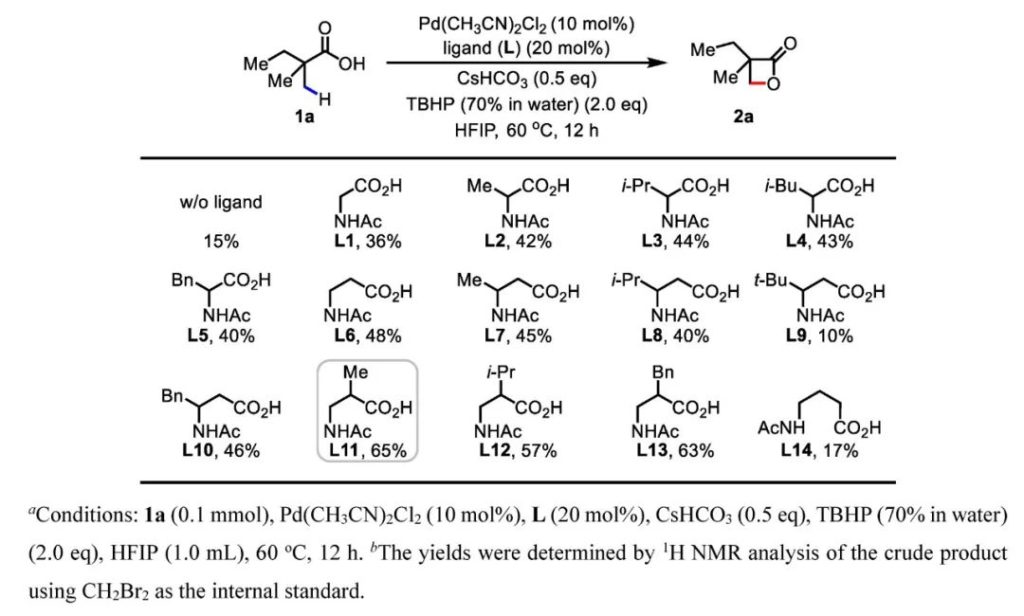 ▲不同配体对反应结果的影响(图片来源:参考资料[3])
▲不同配体对反应结果的影响(图片来源:参考资料[3])
该反应具有良好的官能团兼容性,适用于不同结构烷基羧酸的β-C-H键活化,能以良好至优秀的产率得到一系列β-丙内酯产物。相比于以往发展的C-H键官能化过程,该方法对空气及水汽均不敏感,无需无水无氧操作,并使用廉价的TBHP作为氧化剂,反应结束后简单通过水洗、干燥便可得到纯净的产物,无需柱层析分离提纯,因此十分适合大规模的合成,反应效率也不会受到明显影响。
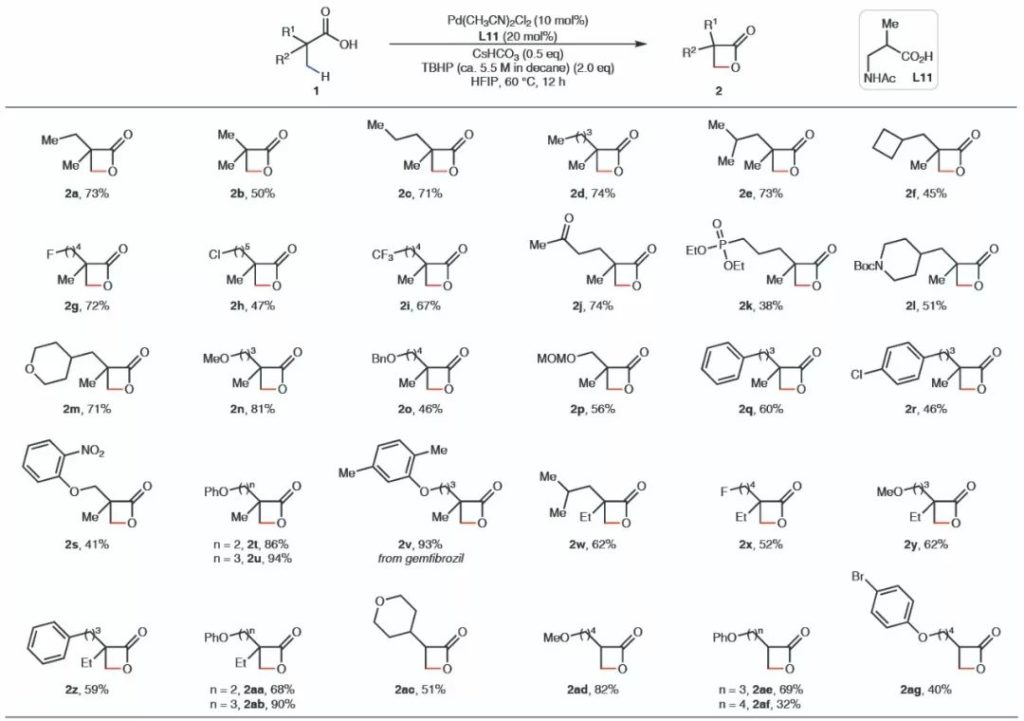
▲合成β-丙内酯中间体的底物适用性(图片来源:参考资料[3])
正如所预料的,β-丙内酯结构具有良好的反应活性,可与一系列C、O、N、S等亲核试剂发生亲核取代开环反应,β-C-O键断裂形成相应的C-C键及C-X键。此前很难引入的二级烷基、富电子的烯基可通过该反应高效地实现,尚无先例的氟化、羟基化及胺化过程也顺利得到解决。由此看来,β-丙内酯确实就像一把“万用刀”,可完成烷基羧酸β-C-H键的多种官能化,具有重要的应用潜力。
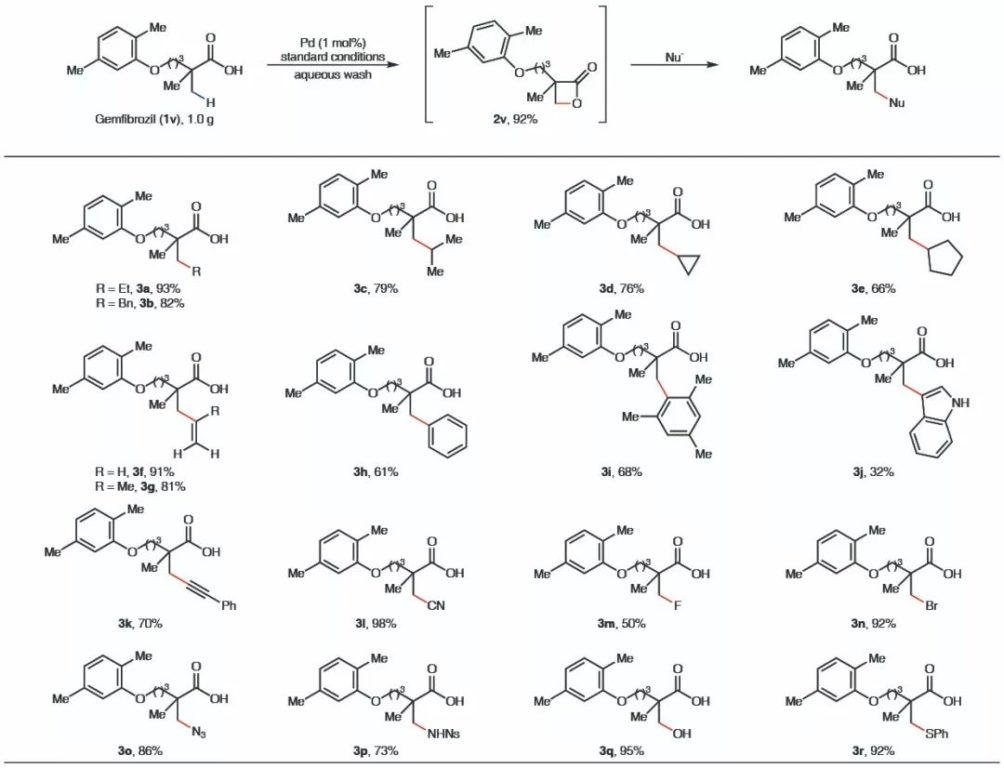 ▲β-丙内酯与一系列亲核试剂的亲核取代开环反应(图片来源:参考资料[3])
▲β-丙内酯与一系列亲核试剂的亲核取代开环反应(图片来源:参考资料[3])
当然,瑞士军刀的智慧不仅体现在一刀多用,还包括对复杂繁冗最大程度的简化。一个刀柄所能容纳的基本工具有限,因而选取时必定经过全方位考量,合理保留必需品与常用器械,省去不必要的累赘,如同生活需要适当做减法。
题图来源:Madeline McCurry-Schmidt / TSRI
参考资料
[1] Pascal S. Engl et al., (2019). C−N Cross-Couplings for Site-Selective Late-Stage Diversification via Aryl Sulfonium Salts. J. Am. Chem. Soc., DOI: 10.1021/jacs.9b07323
消息来源:药明康德传媒
(美国华文网 圣地亚哥华文网编发 USChinesePress.com SanDiegoChinesePress.com)